ENFERMEDADES DEBIDAS A MUTACIONES EN EL CROMOSOMA 15
locus
Enfermedad
|
ENFERMEDADES POR FALLAS EN LOS MECANISMOS DE REPARACIÓN DEL ADN archivo del portal de recursos para estudiantes |
enlace de origen
En la especie humana, las anomalías cromosómicas aumentan con la edad. Sin embargo, se conocen algunas enfermedades, con herencia autosómica recesiva, caracterizadas por la elevada frecuencia de alteraciones cromosómicas en edades tempranas. Estas enfermedades son las conocidas como Síndromes de Inestabilidad Cromosómica, e implican alteraciones en los mecanismos de reparación del DNA. Entre ellos se encuentran el Síndrome de Bloom, la Anemia de Fanconi, la Ataxia Telangiectasia,... . Todos estos síndromes, además de una elevada incidencia de lesiones cromosómicas, presentan una alta predisposición al cáncer y una hipersensibilidad a agentes clastogénicos.
Todos tenemos genes que tienen en ellos pequeńos errores o variaciones. La mayoría de las variaciones no causa enfermedades. Estas variaciones que no producen dańos se llaman polimorfismos. Sin embargo, de vez en cuando, las variaciones son lo suficientemente significativas para causar enfermedades, como es el caso de la ataxia. Las variaciones que producen enfermedades se llaman mutaciones.
A pesar de los últimos avances genéticos, las palabras "ataxia" y "atáxico" son enormemente desconocidas por la sociedad Hispanohablante. "Ataxia" es un vocablo que proviene de una palabra griega que significa “sin orden”. Actualmente la palabra ataxia la conocemos como incoordinación, refiriéndose así a un síntoma. Sin embargo, la palabra "ataxia" se suele utilizar para referirse a un grupo de desórdenes.
Muchas formas de ataxia son genéticas: significa que son causadas por un defecto en un determinado gen. Las enfermedades genéticas, o hereditarias, no son contagiosas, se transmiten de padres a hijos a través de los genes mediante los óvulos o las células de esperma.
Características de las enfermedades autosómicas recesivas:
A - Afectan con igual número de probabilidades a varones y a hembras.
B - Para aparecer los síntomas de la enfermedad deben estar presentes dos copias del gen defectuoso causante de la ataxia .
C - Los portadores de un solo gen de la enfermedad son individuos generalmente normales y sanos, pero pueden transmitir a sus hijos el gen mutado causante de la enfermedad. Y podrían incluso tener un hijo afectado si existiese una combinación con otro gen defectuoso del otro padre.
D - Ambos padres deben ser portadores del gen de la enfermedad para tener un hijo afectado: si ambos padres son portadores del gen defectuoso, la probabilidad de que cada uno de sus hijos herede dos copias del gen defectuoso y desarrolle la enfermedad es del 25 por ciento.
Normalmente, en los desórdenes recesivos los síntomas comienzan en la nińez o en la adolescencia en lugar de en la madurez (aunque por razones que aún no están bien esclarecidas, los síntomas no están necesariamente presentes en el nacimiento o en la infancia).
El síndrome de Bloom es una enfermedad hereditaria rara caracterizada por talla corta, telangiectasia facial (pequeńos vasos sanguíneos faciales dilatados), fotosensitibilidad (sensibilidad creciente a la luz), y susceptibilidad a las infecciones. A lo largo de la vida algunos individuos con síndrome de Bloom tiene riesgo progresivo de malignización. El síndrome de Bloom se hereda como un rasgo genético autosómico recesivo.
La anemia de Fanconi es una enfermedad genética rara que puede presentarse en el nacimiento o durante la nińez. La enfermedad se caracteriza por la pérdida de todos los elementos de la médula ósea, incluyendo los eritrocitos (glóbulos rojos), los leucocitos (glóbulos blancos) y las plaquetas.
Esto dificulta la lucha contra las infecciones, la fatiga y las hemorragias.
La Anemia de Fanconi puede ocurrir en cualquier grupo étnico y nacionalidad, pero cualquier cambio en el gen es responsable por aproximadamente el 83% de los casos de anemia de Fanconi en los descendientes de los Judíos del Este de Europa (Judíos Ashkenazi). Esto permite a los Ashkenazi a ser tratados frecuentemente por este tipo de anemia. Aproximadamente uno en 89 judíos Ashkenazi es portador de la anemia de Fanconi y son capaces de transmitirla a sus hijos si el otro padres es tambien portador.
ATAXIA TELANGIECTASIA
żQUÉ ES LA ATAXIA-TELANGIECTASIA?
Una Enfermedad Multi-sistema: La Ataxia Telangiectasia, o "A-T," es una enfermedad progresiva, degenerativa que afecta a una sorprendente variedad de sistemas del cuerpo. Los nińos con A-T parecen normales en el nacimiento, y las primeras seńales de la enfermedad generalmente aparecen durante el segundo ańo de vida. Estas primeras seńales normalmente son un "tambaleo" o falta de equilibrio y deterioro en el habla causado por la "ataxia", la cual conlleva una pérdida del control muscular.
Ataxia: El inicio de esta ataxia marca el comienzo de la degeneración progresiva de una parte del cerebro, conocida como cerebelo, que gradualmente lleva a una pérdida general del control muscular, y en el futuro confina al paciente a una silla de ruedas. Debido al empeoramiento de la ataxia, los nińos con A-T pierden su habilidad para escribir, y el habla también se retrasa y deteriora. En el futuro, leer se torna imposible porque los movimientos del ojo se vuelven difíciles de controlar.
Telangiectasia: Poco después del inicio de la ataxia, normalmente el paciente muestra otra característica clínica de A-T: "telangiectasias", o las "telarańas" rojas de diminutas venas que aparecen en las esquinas de los ojos o en la superficie de las orejas y en las mejillas expuestas a la luz solar. Aunque estas telangiectasias son inofensivas, su sola aparición junto con la ataxia es lo que llevó a nominar a esta enfermedad como "Ataxia Telangiectasia".
Problemas del sistema de inmunidad: Para la mayoría (aproximadamente 70 por ciento) de los nińos con A-T, hay otra característica clínica: inmunodeficiencia, que normalmente trae infecciones respiratorias reincidentes. En muchos pacientes, estas infecciones pueden tornarse muy graves. Debido a los niveles deficientes en IgA y inmunoglobulina IgE, la infección natural pugna con los representantes en la sangre, los nińos con A-T son muy susceptibles a infecciones pulmonares que no responden a los tratamientos de antibióticos típicos. En éstos pacientes, la combinación de un sistema inmunológico debilitado y la ataxia progresiva pueden llevar finalmente a una pulmonía como una causa corriente de muerte.
Predisposición al cáncer: Frecuentemente, los nińos con A-T tienden a desarrollar malignidades del sistema sanguíneo casi 1.000 veces más que la población general. Particularmente, los linfomas y las leucemias son los tipos corrientes de cáncer, aunque las frecuencias de la mayoría de los cánceres son elevadas. Paradójicamente, otra faceta de la enfermedad es una extrema sensibilidad a la radiación, lo cual significa que los pacientes con A-T normalmente no pueden tolerar la radiación terapéutica dada a los pacientes de cáncer.
Otras características de A-T: Otras características de la Ataxia Telangiectasia que pueden afectar a algunos pacientes son: diabetes mellitus apacible, encanecido prematuro del cabello, dificultad para tragar que causa ahogo y/o babeo, y retraso en el crecimiento. Aunque A-T es un desorden multi-sistema, los nińos afectados tienen y mantienen una inteligencia normal o incluso superior a lo normal. Las conveniencias parecen seguir siendo ayudarles a mantener una perspectiva saludable de vida a pesar de la progresión de sus invalideces.
żQué frecuencia tiene A-T?: La Ataxia Telangiectasia no tiene aspectos raciales, económicos, geográficos, ni barreras de educación. Ambos, varones y hembras son afectados por igual. Los epidemiologistas estiman que la frecuencia de A-T está en una gama entre 1 por 40.000 nacimientos y 1 por 100.000 nacimientos. Pero se cree que nunca serán diagnosticados tantos nińos con A-T como existen, particularmente no lo serán quienes mueren a una edad muy joven. Por consiguiente, realmente esta enfermedad puede ser mucho más corriente.
Prognosis: Actualmente, A-T es incurable y persistente. Si los pacientes tienen la suficiente suerte de no desarrollar cáncer, la mayoría de los nińos A-T dependen de silla de ruedas hacia la edad de diez ańos, no porque sus músculos sean demasiado débiles, sino porque no pueden controlarlos. Después, normalmente los pacientes de A-T mueren de fallo respiratorio o de cáncer en su adolescencia o hacia la década de sus veinte ańos. Algunos pacientes de A-T sobreviven más de los treinta ańos, pero estos casos son sumamente raros.
żQué tratamientos están disponibles?: No hay ninguna cura para A-T, y actualmente no existe ninguna forma de retardar la progresión de la enfermedad. En este momento, los tratamientos sólo se dirigen a aliviar parcialmente algunos síntomas cuando sobrevienen. Porque A-T es rara (enfermedad "olvidada"), los datos de la investigación que están disponibles en terapias farmacéuticas que pueden ayudar a estos nińos, son muy pocos. Física y profesionalmente, la terapia del habla se utiliza para ayudar a mantener la flexibilidad, las inyecciones de la gamma-globulina ayudan a suplementar a los sistemas inmunológicos de los pacientes de A-T, y están proyectándose regímenes de vitaminas a altas dosis con resultados moderados.
La ataxia puede ser un síntoma de muchas enfermedades, incluidos algunos desórdenes asociados con ciertas toxinas, infecciones, y cambios degenerativos en el sistema nervioso. La palabra ataxia puede utilizarse para referirse a una coordinación pobre como síntoma, o puede usarse para denominar una enfermedad degenerativa concreta del sistema nervioso.
Algunos tipos de Ataxia son hereditarios, es decir: son transmitidos por los progenitores. En cambio, otros tipos de Ataxia no parecen tener ninguna conexión hereditaria. En ocasiones, un tipo de Ataxia puede distinguirse de otro por los síntomas. Todos los tipos de Ataxia causan ataxia como síntoma (coordinación pobre). Algunas formas también producen otros síntomas adicionales que las hacen distinguirse de otras variedades con facilidad. A menudo, si los síntomas son apacibles, un médico tiene dificultades para determinar el tipo de Ataxia que tiene el paciente.
Ataxia hereditaria es una alusión a un grupo de enfermedades heredadas que tienen en común el hecho de producir degeneración del cerebelo o de sus vías. El cerebelo es el centro de coordinación del cerebro, por ello, cualquier enfermedad que afecte al cerebelo puede causar una pérdida de coordinación. La coordinación es particularmente importante para caminar y para realizar movimientos con piernas, brazos, manos, lengua, labios, ojos, etc., y funciones como hablar, tragar, etc. Así, frecuentemente, los afectados por desórdenes que afectan al cerebelo notan un equilibrio pobre al caminar, incapacidad para correr, torpeza en las manos, un cambio en su forma de hablar, o movimientos anormales en los ojos.
En los ańos ochenta, fueron desarrollados nuevos métodos para obtener imágenes del cerebro ayudando a determinar la naturaleza cerebral y la situación de los desórdenes del sistema nervioso. La tomografía informatizada y las imágenes por resonancia magnética pueden mostrar si el cerebelo o las partes cercanas al cerebro o el cordón espinal han sido afectadas por un golpe, tumor, infección, o por una enfermedad degenerativa.
Los mayores avances en la investigación de la Ataxia en los ańos noventa han sido el descubrimiento de varios genes causantes de Ataxia. Las pruebas genéticas y las refinadas técnicas para obtener imágenes del cerebro permiten a los médicos hacer diagnósticos a los pacientes de ataxia más exactos y específicos de cuanto era posible hacerlo en el pasado.
Quizás, el mayor grupo de personas afectadas por ataxia es el de quienes notan sus primeros síntomas en la edad adulta, los cuales están desprevenidos por no saber de nadie en la familia afectado por un desorden similar. Esto se llama ataxia esporádica. Para los médicos, estos pacientes son los más difíciles de diagnosticar correctamente, porque hay un gran número de causas de ataxia, adquiridas y hereditarias, que han de ser descartadas antes de poder realizar un diagnóstico de ataxia esporádica con confianza.
Es importante comprender que al diagnóstico de la ataxia esporádica no puede llegarse rápidamente o simplemente por un examen en una visita a la consulta. Dependiendo de la situación, el médico puede necesitar descartar otras causas de ataxia, como un golpe, un tumor cerebral o un cáncer en otra parte del cuerpo, malformaciones congénitas del cerebro, deficiencias de vitaminas, o exposiciones a ciertas toxinas. Si existiese un antecedente significativo de abuso de alcohol, en muchos casos sería imposible determinar si la ataxia es debida a los efectos tóxicos de alcohol en el cerebelo o en los nervios periféricos o se trata de otros desórdenes. Así, antes de recibir un diagnóstico, un paciente debe esperar a los resultados de varios análisis de sangre y otras pruebas del cerebro, del cordón espinal, o una electromiografía.
Otra dificultad para los pacientes con ataxia esporádica y para sus médicos es qué decirles a los parientes. Sería imposible afirmar si una persona con ataxia esporádica tiene una enfermedad hereditaria que simplemente no se ha declarado en otros miembros familiares, o se trata de un estado causado por factores desconocidos en el ambiente.
Otros factores que dificultan el diagnóstico de la ataxia esporádica como enfermedad hereditaria, incluyen la muerte temprana de individuos clave, o falta de comunicación entre parientes, o razones debidas a una adopción. Ocasionalmente, una enfermedad hereditaria puede ocurrir por primera vez en una familia debido a un nuevo cambio o mutación en un gen. El gen responsable de la ataxia telangiectasia fue identificado en 1995 por un equipo de investigadores en Israel.
ATAXIA DE FRIEDREICH (FA)
Es otro tipo de Ataxia hereditaria de tipo temprano. Es una enfermedad heredada que ocasiona dańo progresivo al sistema nervioso ocasionando síntomas que oscilan entre debilidad muscular y problemas de dicción, por un lado, y enfermedad cardiaca, por el otro. Se le llama por el nombre del médico Nicholas Friedreich, que describió inicialmente la condición en la década de 1860. "Ataxia", que se refiere a los problemas de coordinación, tales como movimientos torpes y falta de estabilidad, ocurre en muchas enfermedades y condiciones diferentes. En la ataxia de Friedreich, la ataxia resulta en la degeneración del tejido nervioso en la médula espinal y de los nervios que controlan los movimientos musculares en los brazos y en las piernas. La médula espinal se hace más delgada y las células nerviosas pierden algunos de sus cubrimientos de mielina - la capa de aislamiento en todas las células nerviosas que ayuda a transmitir los impulsos nerviosos.
La ataxia de Friedreich, aunque rara, es la ataxia heredada más prevaleciente que afecta a una de cada 50,000 personas en los Estados Unidos. Hombres y mujeres se ven igualmente afectados.
żCuáles son los signos y síntomas?
Los síntomas comienzan comúnmente entre las edades de 5 y 15 ańos, pero pueden en raras ocasiones aparecer tan pronto como a los 18 meses o tan tarde como a los 30 ańos de edad. El primer síntoma en aparecer es generalmente la dificultad en caminar o ataxia del caminar. La ataxia empeora gradualmente y se propaga lentamente a los brazos y, luego, al tronco. Los signos iniciales incluyen deformidades de los pies tales como pie en forma de porra, flexión de los dedos de los pies (movimientos consistentes en doblar los dedos de los pies involuntariamente), dedos gruesos de los pies en forma de martillo e inversión de los pies (desvío hacia adentro). En el curso del tiempo, los músculos comienzan a debilitarse y a atrofiarse, en especial en los pies, las partes inferiores de las piernas y las manos y aparecen deformidades. Otros síntomas incluyen pérdida de reflejos de los tendones, en especial en las rodillas y en las muńecas y los tobillos. A menudo hay una pérdida paulatina de sensación en las extremidades, que puede propagarse a otras partes del cuerpo. Aparece disartria (lentitud en la dicción o dicción indistinta) y la persona se cansa con facilidad. Son comunes los movimientos rítmicos, rápidos e involuntarios del globo ocular (nystagmus). La mayoría de las personas con ataxia de Friedreich adquieren escoliosis (encurvamiento de la espina dorsal hacia un lado) que, si es aguda, puede dificultar la acción de respirar.
Entre otros de los síntomas que pueden ocurrir figuran dolores de pecho, falta de respiración y palpitaciones cardíacas. Estos síntomas son el resultado de distintas formas de enfermedad cardiaca que a menudo acompańan a la ataxia de Friedreich, tales como la cardiomiopatía (alargamiento del corazón), la miocarditis (inflamación de las paredes del corazón), la fibrosis miocardial (formación de material parecido a fibras en los músculos del corazón) y el fallo cardíaco. También son comunes anomalías rítmicas del corazón tales como taquicardia (latir rápido del corazón) y bloqueo del corazón (conducción dificultada de los impulsos cardíacos dentro del corazón). 20 por ciento, aproximadamente, de las personas con ataxia de Friedreich adquieren intolerancia a los carbohidratos y 10 por ciento de ellas adquiere diabetes mellitus. Algunas personas pierden la capacidad de oír o de ver.
La manera en que progresa la enfermedad varía de una persona a otra. Por lo general, dentro de 15 a 20 ańos después de aparecer los primeros síntomas, la persona queda recluida en una silla de ruedas y, en las etapas posteriores de la enfermedad, las personas quedan totalmente incapacitadas. La expectativa de vida se ve grandemente afectada y la mayoría de las personas con Ataxia de Friedreich mueren en los primeros ańos de la vida adulta, si existe además enfermedad cardiaca seria, la causa más común de la muerte. No obstante, algunas personas con síntomas menos agudos de Ataxia de Friedreich viven por mucho más tiempo.
żCómo se diagnostica la Ataxia de Friedreich?
Los médicos diagnostican la Ataxia de Friedreich realizando un cuidadoso examen clínico, que incluye un historial médico y un minucioso examen físico. Las pruebas que pueden realizarse incluyen lo siguiente:
electromiograma (EMG), que mide la actividad eléctrica de las células musculares,
estudio de conducción de los nervios, que mide la velocidad a la que los nervios transmiten los impulsos,
electrocardiograma (EKG), que da una presentación gráfica de la actividad eléctrica o patrón de pulsaciones del corazón,
ecocardiograma, que registra la posición y movimiento del músculo del corazón,
exploraciones de imágenes de resonancia magnética (MRI) o tomografía computarizada (CT), que proporciona una imagen del cerebro y la médula espinal,
derivación o punción espinal para evaluar el líquido cerebroespinal,
pruebas de sangre y orina para evaluar los niveles elevados de glucosa, y
pruebas genéticas para identificar el gen afectado.
żCómo se hereda la ataxia de Friedreich?
La Ataxia de Friedreich es una enfermedad recesiva autosomal, que significa que el paciente ha de heredar dos genes afectados, uno de cada uno de los padres, para que la enfermedad se desarrolle. Una persona que tiene sólo una copia anormal de un gen para una enfermedad genética recesiva, tal como la ataxia de Friedreich se denomina portador. Un portador no adquirirá la enfermedad pero podría transmitir el gen afectado a sus hijos.
Estos mensajes codificados son "recetas" para fabricar aminoácidos, los bloques de construcción de proteínas. Al combinarse en secuencias largas, como los números de teléfonos largos, las bases "apareadas" indican a cada célula cómo reunir diferentes proteínas. Las proteínas constituyen las células, los tejidos y los enzimas especializados que nuestros cuerpos necesitan para funcionar normalmente. La proteína que se ve alterada en la Ataxia de Friedreich se llama frataxina.
En 1996, un grupo internacional de científicos identificó la causa de la Ataxia de Friedreich como un defecto en un gen situado en el cromosoma 9. Debido al código anormal heredado, una secuencia en particular de bases (GAA) se repite demasiadas veces. Normalmente, la secuencia GAA se repite de 7 a 22 veces, pero en las personas con la Ataxia de Friedreich, ésta se repite de 800 a 1,000 veces. Ese tipo de anormalidad se denomina expansión de repetición triple y ha sido implicada como la causa de varias enfermedades dominantemente heredadas. La Ataxia de Friedreich es la primera enfermedad genética recesiva conocida que es ocasionada por la expansión de repetición triple. Aunque un 98 por ciento de los portadores de Ataxia de Friedreich tienen una expansión triple repetida genética en particular, ésta no se encuentra en todos los casos de la enfermedad. Una proporción muy pequeńa de personas afectadas tienen otros defectos de codificación genética que son responsables de la enfermedad.
La expansión triple repetida perturba aparentemente la conversión normal de aminoácidos a proteína, reduciendo enormemente la cantidad de frataxina que se produce. La investigación indica que sin un nivel normal de frataxina, ciertas células en el cuerpo (especialmente las células del cerebro, de la médula espinal y de los músculos), no pueden soportar cantidades normales de "tensión oxidativa" que producen las mitocondrias, las plantas productoras de energía de las células. Esta indicación sobre la posible causa de la Ataxia de Friedreich se descubrió después de que los científicos realizasen estudios utilizando una proteína de levadura con una estructura similar a la de la frataxina humana. Encontraron que la falta de esta proteína en la célula de levadura conducía a una concentración tóxica de hierro en las mitocondrias celulares. Cuando el excedente de hierro reaccionaba con el oxígeno, se producían radicales libres. Aunque los radicales libres son moléculas esenciales en el metabolismo del cuerpo, pueden también destruir las células y ocasionar dańo al cuerpo. Continúa la investigación en relación a esta materia (véase la sección en este documento "Qué investigación se está realizando?").
żPuede curarse o tratarse la Ataxia de Friedreich?
Al igual que muchas enfermedades degenerativas del sistema nervioso, actualmente no hay una cura eficaz o tratamiento para la Ataxia de Friedreich. Sin embargo, muchos de los síntomas y complicaciones asociados a esta enfermedad pueden ser tratados con el fin de ayudar a los pacientes a mantener un funcionamiento óptimo por el mayor tiempo que sea posible. La diabetes, si se halla presente, puede tratarse con dieta y medicamentos tales como la insulina, y algunos de los problemas cardíacos pueden tratarse con medicamentos también. Los problemas ortopédicos tales como las deformaciones de los pies y la escoliosis pueden tratarse con soportes o cirugía. La terapia física puede prolongar el uso de los brazos y las piernas. Los científicos esperan que los adelantos recientes en la comprensión de los aspectos genéticos asociados a la Ataxia de Friedreich puedan conducir a avances trascendentales en el tratamiento.
żQué servicios son útiles para los pacientes de la Ataxia de Friedreich y para sus familias?
Se dispone de pruebas genéticas en algunos laboratorios especializados y pueden ayudar al diagnóstico clínico, al diagnóstico prenatal y a determinar si el individuo es un portador de la enfermedad. Los asesores genéticos pueden ayudar a explicar cómo se hereda la Ataxia de Friedreich y cuáles son los efectos sobre los pacientes y las familias. El asesoramiento psicológico y los grupos de apoyo para las personas con enfermedades genéticas también pueden ayudar a los pacientes y a sus familias a hacerle frente a la enfermedad.
żQué investigación se está realizando?
En el gobierno federal, el Instituto Nacional de Trastornos Neurológicos y Apoplegía (NINDS- National Institute of Neurological Disorders and Stroke), uno de los componentes de los Institutos Nacionales de Salud (NIH), tiene la responsabilidad principal de patrocinar la investigación sobre los desórdenes neurológicos. Como parte de esta misión, el NINDS realiza investigación sobre la Ataxia de Friedreich y otras formas de ataxias heredadas en sus instalaciones de NIH y también apoya estudios adicionales en centros médicos de todo los Estados Unidos.
Los investigadores se muestran optimistas de que pronto estarán más cerca de comprender las causas de la enfermedad, lo cual facilitará el diagnóstico de los pacientes, asistirá a quienes asesoran a las familias y, con el tiempo, ayudará a los científicos a crear tratamientos eficaces y estrategias de prevención para la Ataxia de Friedreich.
Los estudios con proteínas de levaduras que tienen una estructura química similar a la frataxina humana (véase la sección en este documento "żCómo se hereda la Ataxia de Friedreich?") condujeron a estudios adicionales en ratones y en seres humanos. Estos estudios pusieron de relieve que la frataxina - como la proteína de la levadura - es una proteína mitocondriaca que debería hallarse presente normalmente en el sistema nervioso, el corazón y el páncreas. Sin embargo, en los pacientes con la enfermedad, la cantidad de frataxina en las células afectadas de estos tejidos se ve seriamente reducida. Pruebas adicionales de que la frataxina puede funcionar análogamente a la proteína de levadura fue el resultado de niveles anormalmente elevados de hierro en el tejido del corazón de las personas con Ataxia de Friedreich. Se cree que el sistema nervioso, el corazón y el páncreas pueden ser especialmente susceptibles a dańos por radicales libres (producidos cuando el exceso de hierro reacciona con el oxígeno), debido a que una vez que ciertas células en estos tejidos quedan destruidas por los radicales libres no pueden ser reemplazadas. Las células de los nervios y de los músculos también tienen necesidades metabólicas que pueden hacerlas especialmente vulnerables a dańos por los radicales libres. Los radicales libres han sido implicados en otras enfermedades degenerativas, tales como las enfermedades de Parkinson y de Alzheimer.
Armados con lo que actualmente conocen acerca de la frataxina y la ataxia de Friedreich, los científicos trabajan por definir mejor el papel de la frataxina, aclarar la forma en la que los defectos en el metabolismo del hierro pueden desempeńar un papel en el proceso de la enfermedad y explorar nuevos enfoques terapéuticos para la enfermedad. El descubrimiento por investigadores apoyados por NINDS de la mutación genética que ocasiona la Ataxia de Friedreich ha dado nuevo ímpetu a las actividades de investigación de esta enfermedad.
SÍNDROME DE BLOOM
żQUÉ ES EL SÍNDROME DE BLOOM?
Este síndrome está caracterizado por eritema facial con telangiectasias, estatura baja, respuesta inmune anormal y predisposición a tumores. Se hereda de manera autosómica recesiva.
Se han realizado estudios cromosómicos en fibroblastos y linfocitos en cultivo, que han mostrado aberraciones cromosómicas, con intercambios anormales de cromátides hermanas, aumento de la sensibilidad a la RUV y alteraciones en la síntesis de ADN. Se ha demostrado actividad anormal del ADN ligasa.
Clínicamente, el paciente tiene una facies característica, con un rostro alargado, facciones delicadas, nariz prominente y eritema, que se manifiesta como máculas o placas eritematosas con descamación fina en mejillas, puede también afectar la frente, los pabellones auriculares y otras áreas fotoexpuestas. Con la exposición al sol, las lesiones se exacerban y pueden mostrar la aparición de ampollas Estos nińos son bajos, delgados, de huesos finos. Los cambios en la anatomía craneofacial y el paladar ojival condicionan un timbre de voz agudo.
El síndrome se asocia con frecuencia con manchas café con leche, sindactilia, malformaciones cardíacas, páncreas anular y muchos otros defectos congénitos.
Muchos hombres con el síndrome son estériles por atrofia de los túbulos seminíferos; sin embargo, la función endocrina testicular parece preservarse.
Aparentemente, estos nińos tienen inteligencia normal; sin embargo, muchos tienen problemas de aprendizaje por su apariencia física con alteraciones de la personalidad.
Estos pacientes están predispuestos a neoplasias, particularmente leucemias.
Se han descrito pocos casos. Afecta principalmente a varones. Muchos de estos pacientes son descendientes de judíos Ashkenazi en los que la incidencia es de 1/160000.
Presentan bajo peso al nacer y retraso en el crecimiento. Desarrollan eritemas en la piel al exponerse al sol y problemas inmunológicos. Presentan elevada predisposición al cáncer, especialmente leucemias y limfomas.
Citogenéticamente, muestran varias características:
1·elevada frecuencia de roturas y reorganizaciones cromosómicas
2·presencia de intercambios "simétricos" cuatrirradiales y de intercambios entre cromosomas homólogos.
3·frecuencia elevada de intercambio de cromátides hermanas (característico de este síndrome)
Se debe a una mutación del gen BS (BSM), situado en el cromosoma 15q26.1, que codifica para la RecQ helicasa una enzima que se requiere para la replicación y reparación del DNA. Todas las mutaciones de pacientes Ashkenazi son idénticas, indicando un origen común con una subsiguiente expansión de esta mutación a la población.
La mutaciones que causan el Síndrome de Bloom borran o alteran las funciones de la Helicasa y puede desactivar la actividad de lectura 3’-5’ de ésta.
|
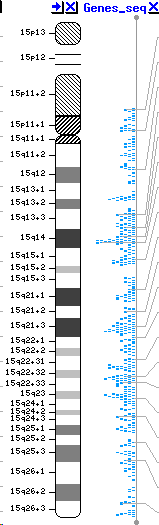
ENFERMEDADES DEBIDAS A MUTACIONES EN EL CROMOSOMA 15 |
||
|
|
locus
|
Enfermedad
|
| 15q11-q13 | Síndrome de Prader-Willi | |
| 15q11-q13 | Albinismo oculocutáneo | |
| 15q11-q13 | Síndrome de Angelman | |
| 15q14 | Esquizofrenia | |
| 15q15 | Sordera por déficit de estereocilina | |
| 15q15 | Esferocitosis | |
| 15q15-q21.1 | Síndrome de Bartter | |
| 15q15.1-q21.1 | Distrofia muscular | |
| 15q15.3 | Hipotiroidismo congénito | |
| 15q21.1 | Ginecomastia familiar | |
| 15q21 | Síndrome de Griscelli | |
| 15q21.1 | Síndrome de Marfan | |
| 15q22.1 | Cardiomiopatía hipertrófica familiar | |
| 15q22.31-15q25.3 | Enfermedad de Tay-Sachs | |
| 15q22.3-q23 | Síndrome de Bardett-Biedl | |
| 15q22.3-q23 | Artritis piogénica estéril | |
| 15q23-q25 | Tirosinemia, tipo I | |
| 15q25 | Oftalmoplegia progresiva | |
| 15q26.1 | Síndrome de Bloom | |
SÍNDROME DE COCKAYNE
Esta es una enfermedad rara que, en contraste con el síndrome de Bloom, se caracteriza por retraso mental, que se asocia a fotosensibilidad, estatura baja, grandes pies, manos y orejas, así como encefalopatía y neuropatía periférica condesmielinización.
La herencia es de tipo autosómico recesivo. Los fibroblastos crecen mal en cultivo y muestran intensa sensibilidad a la RUV. Se ha demostrado que la síntesis de ADN y ARN y fibroblastos disminuye después de la radiación.
Clínicamente, el nińo en general no muestra anormalidades durante el primer ańo de vida. Sin embargo, más tarde hay eritema marcado después de exposición al sol, el cual evoluciona hacia manchas hiper o hipopigmentadas y cicatrices atróficas en áreas fotoexpuestas. Estos cambios resultan en un aspecto prematuramente envejecido, lo que se acentúa por la atrofia del tejido celular subcutáneo facial y orbitario, así como por el enrarecimiento y encanecimiento prematuro del cabello. La cara fina, asociada a orejas, manos y pies grandes sugieren una similitud a la familiar figura del ratón Miguelito.
El retraso mental de estos pacientes se asocia a extensas áreas de desmielinización del sistema nervioso central y periférico. También hay retardo del crecimiento físico y ceguera por atrofia óptica y cataratas. La muerte ocurre, en la mayoría de los casos, entre los 15 y 20 ańos.
El diagnóstico diferencial se debe hacer con la progeria, en la cual no hay hipersensibilidad a la luz, alteraciones oculares y capilares, ni el gran tamańo relativo de las orejas, manos y pies. En el síndrome de Bloom no ocurre retraso mental y en el xeroderma pigmentoso no se observa desmielinización.
ANEMIA DE FANCONI
żQUÉ ES LA ANEMIA DE FANCONI?
La Anemia de Fanconi es una enfermedad hematológica rara, caracterizada por el desarrollo gradual de pancitopenia (disminución de las tres series celulares sanguíneas a la vez, hematíes o glóbulos rojos, leucocitos o glóbulos blancos y plaquetas), durante la infancia, presentándose con más frecuencia en los varones con una proporción 1,3:1. Se asocia con frecuencia a diversas anomalías congénitas: anomalías del esqueleto, corazón y rińones, malformaciones del sistema nervioso central (sistema formado por el encéfalo y la médula espinal) con retraso mental y pigmentación anormal de la piel.
Fue descrita por Fanconi en 1927 y en 1931 Naegeli sugirió que el término Anemia de Fanconi se debía utilizar para la anemia aplásica (disminución de los hematíes o glóbulos rojos circulantes por la insuficiencia de todos los elementos formes de la sangre como consecuencia de que la médula ósea no es capaz de generar nuevas células) familiar con malformaciones congénitas asociadas.
El cuadro clínico suele ser asintomático durante la primera infancia, cuatro a siete ańos en los nińos y seis a diez ańos en las nińas, en que los pacientes comienzan con hemorragias, infecciones o clínica de anemia (disminución de los hematíes o glóbulos rojos circulantes), según predomine la trombopenia (disminución de las plaquetas circulantes, y que intervienen en la coagulación de la sangre), neutropenia (niveles anormalmente bajos de neutrófilos, un tipo de células blancas de la sangre) o anemia.
La sintomatología progresa de forma gradual y los síntomas se deben al desarrollo de una pancitopenia progresiva. Inicialmente se presentan las manifestaciones clínicas atribuibles a la trombocitopenia: petequias (manchas pequeńas en la piel, formadas por la salida de sangre), hematomas (colección sanguínea enquistada), episodios graves de epistaxis (hemorragias nasales) y sangrado gastrointestinal; más tarde se evidencian los signos de anemia: palidez, fatiga fácil, debilidad e hiporexia (disminución del apetito).
El 75% de los pacientes presentan anomalías congénitas asociadas:
1. Retraso del crecimiento, en algunos nińos se ha encontrado deficiencia en la producción de la hormona del crecimiento. La asociación de alteraciones del metabolismo de la melanina y el retraso del crecimiento, parece sugerir un defecto en el área hipotálamo hipofisaria que controla la secreción de estas hormonas
2. Hiperpigmentación cutánea: de distribución central que afecta al cuello, axilas, areola mamaria, abdomen, ombligo, genitales e ingles. También puede presentarse en forma difusa, con predilección por los pliegues cutáneos o aparecer en forma de manchas café con leche. La lesión histológica (histología es la parte de la anatomía que estudia los tejidos que forman los seres vivos) corresponde esencialmente a hipermelanosis (aumento anormal de la melanina, pigmento marrón oscuro o negro que existe en el pelo, la piel, el iris y la coroides del ojo)
3. Alteraciones del sistema nervioso central y órganos de los sentidos: microcefalia (cabeza anormalmente pequeńa), microftalmía (ojos anormalmente pequeńos), epicantus (dobleces adicionales de la piel en las esquinas internas de los ojos), ptosis palpebral (párpados caídos), estrabismo (desviación de uno de los ojos de su dirección normal, por lo que los ejes visuales no pueden dirigirse en un mismo tiempo al mismo punto), nistagmus (espasmos de los músculos del ojo que produce movimientos oculares rápidos e involuntarios), sordera y deformaciones del pabellón auricular
4. Anomalías esqueléticas, presentes en el 75% de los casos, especialmente del miembro superior. Pueden presentar anomalías en antebrazos como aplasia (ausencia de desarrollo) o alteraciones morfológicas del radio, en las manos como hipoplasia (desarrollo incompleto o defectuoso) del primer metacarpiano o de la eminencia tenar (elevación de forma redondeada situada en la palma de la mano, cerca de la base del dedo pulgar) y anomalías de los dedos pulgares que van desde ausencia del dedo hasta hipoplasia de la uńa y pulgares trifalángicos.
5. Anomalías renales: duplicación de pelvis, hidronefrosis (acumulo anormal de orina en los rińones), quistes, rińón en herradura o rińones ectópicos (situación fuera de su lugar habitual de un órgano o tejido).
La anemia aplásica de Fanconi, es la causa más frecuente de asociación de pancitopenia con malformaciones congénitas asociadas.
La sospecha diagnóstica viene dada por la presencia de una pancitopenia en el hemograma (resultado del estudio cualitativo y cuantitativo de los elementos formes de la sangre) que se acompańa de macrocitosis (proliferación anormal en la sangre periférica de macrocitos que son eritrocitos o glóbulos rojos maduros de tamańo anormalmente grande). La existencia de pancitopenia en un nińo obliga a la búsqueda de malformaciones físicas asociadas, y a descartar una causa congénita.
El estudio citogenético (estudio a nivel de la célula de los caracteres particulares de la herencia, principalmente de los cromosomas y los genes) de linfocitos (un tipo de leucocitos o glóbulos blancos de la sangre) en sangre periférica es imprescindible para el diagnóstico. La demostración de una fragilidad cromosómica aumentada es característica de la anemia de Fanconi. Es muy frecuente encontrar un aumento de la expresión del antígeno "i" y un aumento de la hemoglobina F, como reflejo de una eritropoyesis (mecanismo por el que se forma la sangre) de stress.
Una proporción importante de pacientes pueden presentar diversas alteraciones cromosómicas: fracturas, translocación de cromátides y endorreduplicaciones en linfocitos, fibroblastos (células procedentes de las células conjuntivas en vías de proliferación) y células de la médula ósea.
El estudio medular, que siempre debe incluir biopsia (operación que consiste en extirpar en el individuo vivo un fragmento de órgano o de tumor con objeto de someterlo a examen microscópico) ósea, demuestra hipoplasia (desarrollo incompleto o defectuoso) de la médula ósea con depresión de las tres líneas celulares.
La anemia es de tipo crónica arregenerativa, generalmente normocítica (anemia con eritrocitos o glóbulos rojos maduros de tamańo normal) o ligeramente macrocítica (anemia con eritrocitos o glóbulos rojos maduros de tamańo aumentado); el porcentaje de reticulocitos (un tipo de glóbulo rojo) es comúnmente bajo. El recuento leucocitario revela habitualmente granulocitopenia (niveles anormalmente bajos de granulocitos, un tipo de células blancas de la sangre), en especial neutropenia (niveles anormalmente bajos de neutrófilos, un tipo de células blancas de la sangre) con linfocitosis (aumento anormal de linfocitos, un tipo de glóbulos blancos, en sangre periférica) relativa.
El tratamiento paliativo se basa en el control de la anemia mediante el uso de transfusiones de plaquetas, eritrocitos o leucocitos; o de estrógenos, que pueden producir efectos secundarios como el desarrollo de leucemias o cáncer de hígado.
El tratamiento curativo de la anemia de Fanconi, es el transplante de médula ósea.
Se hereda como un rasgo genético autosómico recesivo.
NOTICIA NOVEDOSA DE INTERES
Octubre 4 2001
Nace un bebé seleccionado por sus genes para salvar a su hermana
Su embrión fue elegido tras extraer una célula y comprobar que era idóneo para realizar un trasplante a su hermana de seis ańos, afectada por una enfermedad incurable - La operación ha sido realizada con éxito en EEUU
NUEVA YORK.- Una pareja de Colorado (EEUU) se ha convertido en la primera familia del mundo que ha elegido los genes de su hijo. El pequeńo Adam Nash ha nacido para salvar la vida de su hermana Molly, de seis ańos, que necesita sus células sanas para vivir. Su embrión fue seleccionado entre varios tras comprobar que sus células curarían a Molly, que sufre anemia de Fanconi, una enfermedad incurable. Molly recibió un trasplante el pasado día 26 de septiembre. Dentro de una semana, según los médicos, se sabrá si la intervención ha tenido éxito.
Lisa y Jack Nash no buscaban un bebé más alto, más rubio o más inteligente que dentro de 20 ańos sea el más listo de su clase o que rompa los corazones de las chicas con sus facciones perfectas y sus ojos azules. Los Nash han utilizado pruebas genéticas para elegir, entre varios embriones, un hijo que naciera con las células exactas para tratar la enfermedad incurable de su hermana. Es la primera vez que un embrión, destinado a crear un ser humano mediante fertilización in vitro, es sometido a pruebas genéticas para buscar células compatibles con otro ser humano, en este caso, su hermana Molly.
Poco después de nacer, fueron extraídas del cordón umbilical del pequeńo Adam, mediante una intervención sin dolor, las células que su hermana Molly necesita para fortalecer su médula y salvarse de una muerte segura. La nińa sufre de anemia de Fanconi, una enfermedad que afecta al desarrollo de las células en la médula y que produce anemia, hemorragias y desórdenes inmunológicos que suelen producir la muerte a los 7 ańos a causa de leucemia o de otras complicaciones similares.
La familia Nash se sometió cinco veces a una diagnosis de preimplantación genética, el análisis genético del padre y de la madre, para determinar qué células son normales. Cuando se encuentra la célula ideal se realiza la fecundación in vitro con el embrión perfecto -en este caso sin la enfermedad de Fanconi-, que es implantado en el útero de la madre.
El embrión perfecto: «Molly tenía a Adam en sus brazos y, aunque para nosotros fue algo extraordinariamente monumental, parecía tan simple... En un primer momento piensas que va a haber rayos y truenos y que todo va a salir mal, pero fue algo muy tranquilo», recordó ayer Lisa Nash al diario The Washington Post, hablando del momento en que las células del pequeńo Adam entraron por un tubo en el pecho de su hermana Molly.
Este bebé no es el primer nińo que nace para salvar la vida de un familiar. Médicos de todo el mundo llevan décadas intentando evitar la muerte de enfermos de leucemia, y de otras enfermedades hereditarias, gracias a la sangre y algunos órganos vitales de sus hermanos y sus familiares.
La llegada al mundo de Adam, el pasado 29 de agosto en Denver, ha despertado mucho interés en la comunidad médica y ética de Estados Unidos porque es la primera vez que los padres de un bebé examinaron y eligieron el embrión que fue implantado artificialmente en el interior de su madre para que el nińo salvador naciera.
Molly fue intervenida el pasado día 26 de septiembre, momento en que recibió las células sanas de su hermano Adam. Los doctores que cuidan de Molly, de seis ańos de edad, en Minneapolis no sabrán si la transfusión de las células de su hermano tendrá resultados positivos hasta dentro de una semana. De lo que si están seguros es que este trasplante abre una nueva fase en las investigaciones sobre la reproducción humana, su futuro y las consideraciones éticas y morales que rodean la decisión de engendrar un hijo y poder elegir los genes que tendrá.
«De momento, Molly no ha tenido complicaciones. Se ha levantado y está jugando con un ordenador en su habitación», declaró John Wagner, especialista de la Universidad de Minnesota que ha supervisado la intervención. Pronto habrá superado el periodo de alto riesgo que conlleva todo trasplante. Los médicos reforzaron su sistema inmunológico con radiación y quimioterapia para aumentar las posibilidades de que las células no fueran rechazadas.
El diario The Washington Post seńalaba ayer que la selección previa de los embriones para elegir los genes ideales, llevará al descubrimiento de nuevas terapias que salven muchas vidas en peligro. «Sabíamos que se nos estaba acabando el tiempo», ha declarado al diario capitalino Charles Strom, el doctor del Illinois Masonic Medical Center, donde se realizaron las pruebas genéticas para elegir al embrión perfecto.
Strom asegura que las nińa tiene ahora, gracias a las células de su hermano, entre un85a un 90% de posibilidades de vivir sin la enfermedad en su médula. Pero para muchos doctores, la caja de Pandora de la experimentación genética -y no siempre con buenas intenciones- se está abriendo de par en par.
Críticas éticas: «Esto se está convirtiendo rápidamente en algo similar a comprar un coche y poder decidir el paquete de accesorios extra que quieres», ha advertido Jeffrey Kahn, director del centro de bioética de la Universidad de Minnesota. Este experto cree que nos encontramos a la vuelta de la esquina de un futuro en el que los padres pidan y exijan embriones para tener hijos que no sean homosexuales o que midan 1,80 metros de altura. «No lo hacen ahora porque no tenemos los análisis necesarios para poder determinarlo», ha asegurado Kahn al Post.
En un principio, los padres de Molly no querían tener más hijos, ya que tenían miedo de que el futuro nińo estuviera afectado por la misma enfermedad. Tanto Lisa como Jack Nash incorporan en sus genes una versión normal y una versión defectuosa del gen Fanconi y, por lo tanto, tenían un 25% de posibilidades de engendrar un bebé afectado con cada embarazo.
Por esta razón, los médicos optaron por fecundar in vitro varios embriones en lugar de que el matrimonio utilizara el método natural de embarazo. En uno de estos embriones estaba el pequeńo Adam que hoy abrazan, el benjamín que posiblemente salvará la vida de su hermana Molly.
La anemia de Fanconi, entidad autosómica recesiva se caracteriza por pancitopenia, malformaciones congénitas e inestabilidad cromosómica; las manifestaciones clínicas suelen iniciarse a los seis ańos de edad. Se presenta un paciente de 19 meses de edad cuyo diagnóstico se estableció en fase preanémica. Se discuten aspectos génicos y cromosómicos de la entidad y la importancia de establecer el diagnóstico en etapa temprana.
CONCLUSIONES
Cualquiera que alguna vez haya arrojado una moneda al aire, sabe lo que ocurre con ese promedio teórico. Se puede arrojar una moneda seis veces seguidas y se puede sacar "caras" las seis veces, o "caras" cuatro veces y "cruces" dos veces, o al revés. Igualmente, un hombre o mujer que portan un gen causante de una ataxia dominante podrían tener seis nińos que todos reciben el gen defectuoso, o todos el gen normal, o cualquier combinación: en cada nuevo caso interviene el azar.
El cuerpo tiene que copiar 100.000 genes con precisión al realizar cada óvulo o cada célula de esperma. Durante ese proceso de copia suceden errores o cambios, y una vez ocurrido el error en un gen causante de ataxia, la enfermedad puede aparecer por primera vez en la familia. Una vez habida una mutación del gen, puede transmitirse a la próxima generación. Muchos pacientes de ataxia no tienen ninguna historia familiar de ataxia. Es difícil determinar si éstos casos esporádicos de ataxia se deben a alguna causa no genética o representan el primer caso de un desorden genético en la familia. Los especialistas en Ataxia han esperado con avidez a la disponibilidad de las pruebas genéticas para ayudarles a responder esta pregunta a sus pacientes.
Modelo autosómico de herencia recesiva: Autosómico quiere decir que es igualmente probable que las enfermedades recesivas ocurran en varones y en hembras porque los genes responsables se localizan en un par del autosoma (un autosoma es cualquiera de los cromosomas que no son cromosomas del sexo). Difieren de las enfermedades dominantes, en que se ha de tener dos copias del gen defectuoso para padecer los síntomas de la enfermedad. Las personas sólo desarrollan síntomas de la enfermedad cuando ninguna de las dos copias de un gen recesivo trabaje correctamente. Las formas recesivas más corrientes de ataxia son la de Friedreich y la ataxia telangiectasia.
Características de las enfermedades autosómicas recesivas:
A - Afectan con igual número de probabilidades a varones y a hembras.
B - Para aparecer los síntomas de la enfermedad deben estar presentes dos copias del gen defectuoso causante de la ataxia .
C - Los portadores de un solo gen de la enfermedad son individuos generalmente normales y sanos, pero pueden transmitir a sus hijos el gen mutado causante de la enfermedad. Y podrían incluso tener un hijo afectado si existiese una combinación con otro gen defectuoso del otro padre.
D - Ambos padres deben ser portadores del gen de la enfermedad para tener un hijo afectado: si ambos padres son portadores del gen defectuoso, la probabilidad de que cada uno de sus hijos herede dos copias del gen defectuoso y desarrolle la enfermedad es del 25%.
Normalmente, en los desórdenes recesivos los síntomas comienzan en la nińez o en la adolescencia en lugar de en la madurez (aunque por razones que aún no están bien esclarecidas, los síntomas no están necesariamente presentes en el nacimiento o en la infancia).
 Para tener dos copias de un gen defectuoso, un hijo debe recibir una copia del gen de la enfermedad de cada padre. La mayoría de las veces, ningún padre sabe que él/ella tienen un gen defectuoso hasta que aparece un hijo con un desorden recesivo. Incluso entonces, la naturaleza genética del desorden no puede apreciarse, y los síntomas del nińo pueden atribuirse a otras causas. A menudo, la causa genética no es sospechada hasta que aparece un segundo hijo que tiene el mismo desorden.
Para tener dos copias de un gen defectuoso, un hijo debe recibir una copia del gen de la enfermedad de cada padre. La mayoría de las veces, ningún padre sabe que él/ella tienen un gen defectuoso hasta que aparece un hijo con un desorden recesivo. Incluso entonces, la naturaleza genética del desorden no puede apreciarse, y los síntomas del nińo pueden atribuirse a otras causas. A menudo, la causa genética no es sospechada hasta que aparece un segundo hijo que tiene el mismo desorden.
Por ultimo, podemos concluir que estas tres enfermedades principales de las cuales hablamos en el trabajo, se heredan como un rasgo genético autosómico recesivo.
OTRAS ENFERMEDADES RELACIONADAS CON LA FALLA EN LOS MECANISMOS DE REPLICACIÓN
XERODERMA PIGMENTOSA – XP
Un raro desorden genético en el que la radiación ultravioleta ha inducido cambios en los mecanismos de reparación del DNA; caracterizado por severa sensibilidad a todas las fuentes de radiación ultra violeta - UV (especialmente a la luz solar). Se conocen menos de 1000 casos en todo el mundo.
XP está categorizada en grupos de acuerdo a la capacidad del cuerpo para reparar el DNA. Grupos A, C, D y Variantes cubren el 90% de todos los casos de XP. El grupo A, por ejemplo, tiene el menor nivel de reparación y las mayores manifestaciones neurológicas.
Rango variado de síntomas
- ceguera y sordera
- ampollas y pecas con la exposición mínima solar
- incapacidades del desarrollo
- enanismo e hipergonadismo
- incremento de cánceres de piel y de ojos
- retardo mental
Otros sindromes relacionados
- Atáxia-Telangiectasia
- Síndrome de Bloom
- Síndrome de Cockayne
- Anemia de Fanconi
- Trichothiodystrophy (TTD)
Amenaza de vida
- El dańo del DNA es acumulativo e irreversible
- No hay cura para la XP. Su administración esta limitada a evitar la exposición a la danina
radiación UV, mantenerse en interiores a los que se les ha bloqueado la entrada de la luz
solar, y al uso de ropas protectoras, lociones bloqueadoras y anteojos para evitar los rayos
del sol. También evitar otros conocidos carcinogenos.
Chequeos regulares para detectar y tratar neoplasias es muy importante.
BIBLIOGRAFÍA
http://members.nbci.com/hispataxia/ATAXIA/YATE-PP.htm
http://members.nbci.com/hispataxia/ATAXIA/YATX17.htm
http://www.vistamedica.com/enfermed.asp
http://salud.medicinatv.com/consejos/muestra.asp?id_consejo=514
http://cisat.isciii.es/er/prg/er_bus1.asp?letra=A&procedencia=er_busal
http://www.xps.org/Spanish.htm
http://genmic41.uab.es/Biocomputacio/treballs98-99/Sarrate/totbo/ejb.html
http://genmic41.uab.es/Biocomputacio/treballs98-99/Sarrate/totbo/bbb.html